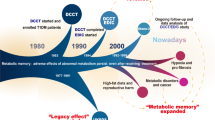
In recent years, several post-interventional analyses of large-scale randomized controlled clinical trials have given us a new concept regarding the risk management of hypertension and cardiovascular diseases. The beneficial effects of intensive treatments were extended even after the interventions ended. This phenomenon is known as “metabolic memory” or “legacy effect”, and we recognized its clinical significance. A certain level of evidence in human and animal studies employing organ transplantation techniques has indicated that this type of “memory” resides in each organ and could be transferrable, erasable, and rewritable, which is similar to neuronal and immune “memory”. In this review, we define this memory as “organ memory” and summarize the current picture and future direction of this concept. “Organ memory” can be observed in many clinical settings, including in the control of hypertension, diabetes mellitus, and dyslipidemia. Several intensive treatments were demonstrated to have the potential to rewrite “organ memory”, leading to the curability of targeted diseases. “Organ memory” is the engraved phenotype of altered organ responsiveness acquired by a time-dependent accumulation of organ stress responses. Not only is the epigenetic change of key genes involved in the formation of “organ memory” but the alteration of multiple factors, including low molecular weight energy metabolites, immune mediators, and tissue structures, is involved as well. These factors intercommunicate during every stress response and carry out incessant remodeling in a certain direction in a spiral fashion through positive feedback mechanisms. Future studies should be directed toward the identification of the core unit of “organ memory” and its manipulation.
In several randomized controlled clinical trials (RCTs), the “memory phenomenon” or “legacy effect” has been proposed by clinical evidence to show that, even after the cessation of the clinical trial, the superiority of one treatment over the other persists. This proposal is now highlighted for its medical significance. The existence of “memory” acquisition by transient interventions has been experienced in many clinical contexts of non-communicable diseases (NCDs) such as lifestyle-related diseases (diabetes mellitus, hypertension, and so on). As described later, we have come to recognize that this kind of “memory” is supposed to reside in each organ, so we designate it “organ memory”.
The actual entity of “organ memory”, however, has barely been examined. “Organ memory” is the engraved phenotype of altered organ responsiveness acquired by time-dependent accumulation of organ stress responses and is expected to have a great potential to offer a new insight into medical science. In this review, we delineate the concept of “organ memory” from multiple aspects including clinical settings, mechanistic insight, and molecular basis.
In clinical settings, the “memory phenomenon” can cause both beneficial and deleterious effects. Previous insufficient risk control was revealed to have persistent effects on the occurrence of lifestyle-related diseases, such as hypertension and diabetes, or the progression of their vascular complications, even though the present control was satisfactory. Past history of obesity, hypertension, hyperglycemia, and smoking is recognized to continuously generate a harmful effect on the occurrence of cardiovascular events. Although it is assured that smoking cessation is beneficial for risk reduction, most large-scale cohort studies have indicated that ex-smokers still have a residual risk of vascular diseases compared with never smokers [1, 2]. As another example, we previously reported that past obesity could influence the progression of diabetic nephropathy even if the current body weight is within the normal range [3].
In contrast, persistent beneficial effects of the transient intervention for preventing cardiovascular events and its related mortality were observed even after completion of the intervention in various large-scale clinical trials. In regard to the RCTs on intensive glucose lowering in type 2 diabetes, a 10-year post-trial follow-up of UKPDS (United Kingdom Prospective Diabetes Study) revealed that a relative reduction of cardiovascular risks in the intensive treatment group lasted long after the intervention ended, although superior glucose lowering by the intensive treatment was lost soon after the trial ended [4]. The DCCT (Diabetes Control and Complications Trial)/EDIC (Epidemiology of Diabetes Interventions and Complications) study demonstrated that intensive therapy in type 1 diabetes significantly slowed the progression of diabetic retinopathy and nephropathy during 6.5 years of the intervention period (DCCT) [5]. The reduction resulting from intensive therapy persisted for at least 4 years of the post-trial follow-up period (EDIC) [6], despite the fact that glycemic control between the intensive and standard group was similar during the post-trial period. For blood pressure control, the ADVANCE (Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation)-ON showed a beneficial post-trial “memory phenomenon” with respect to reductions in the risk of death from cardiovascular causes [7]. In a lipid-lowering study, WOSCOPS (the West of Scotland Coronary Prevention Study) demonstrated that cardiovascular events and related deaths were reduced even during the subsequent 10-year post-trial follow-up period after the 5-year treatment of hypercholesterolemia by pravastatin [8]. The 4 S (the Scandinavian Simvastatin Survival Study) study also showed a “memory phenomenon” by a transient treatment of hypercholesterolemia by simvastatin [9].
In some cases, the beneficial effects of the intensive treatment first became evident during the post-trial follow-up period. In the DCCT/EDIC study, the reduction in major cardiovascular events, which consisted of nonfatal myocardial infarction, nonfatal stroke, and cardiovascular death, was not shown during the intervention period. The significant reduction associated with the intensive treatment became evident during the post-trial period [10]. The ACCORD-BP (Action to Control Cardiovascular Risk in Diabetes blood pressure trial) examined the effect of intensive blood pressure control in high-risk patients with type 2 diabetes. A significant reduction in major cardiovascular events was not observed during the 5 years of the intervention period [11]. The reduction caused by the early intensive treatment became evident in a subgroup analysis of the ACCORD Follow-On trial (ACCORDION) in which ACCORD participants were subsequently followed for 4 years [12].
In addition to the reduction in cardiovascular events, the “memory phenomenon” after a transient intervention has been observed in the onset of hypertension. The TROPHY (the Trial of Preventing Hypertension) study demonstrated that the risk of the progression from the prehypertension stage to hypertension was reduced by a 2-year transient inhibition of the renin–angiotensin system (RAS) by candesartan, and this reduction was observed even after the trial period was finished [13]. We have shown this kind of “memory phenomenon” in hypertension-prone animal models [14,15,16], which we termed as “angiotensin block memory”. In contrast, the activation of RAS in youth has been shown to induce hypertension, even after the cessation of RAS activation [17].
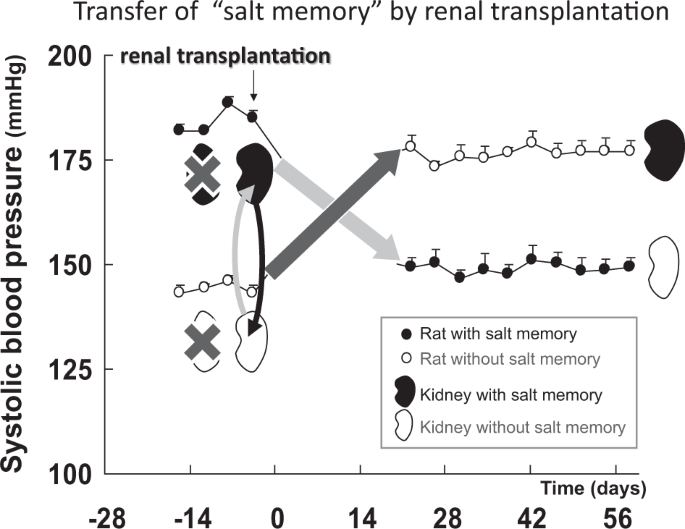
In humans, salt intake in the neonatal stage could determine late-stage blood pressure, which is also considered a “memory phenomena”. A randomized trial was conducted and demonstrated that sodium restriction in newborn infants influenced blood pressure in a 15-year post-trial follow-up [18]. We recently reported that a persistent hypertensive phenotype was acquired by transient high salt intake in two experimental models of hypertension (spontaneously hypertensive rat stroke prone, SHR-SP, and the Dahl salt sensitive hypertensive rat) and proposed this phenotype as “salt memory” [19].
To better understand the characteristics of the “memory phenomenon” in clinical settings, we performed a systematic review of RCTs of cardiovascular diseases that investigated the “memory phenomenon”, namely, the post-trial effects of the transient intervention, as we have reported recently [20]. In that systematic review, “memory phenomenon” was identified as positive when the cardiovascular outcome was significantly superior in the intervention group even at the end of the post-trial observational follow-up period. At that point, the significant reduction of a risk factor (blood glucose, blood pressure, or lipid level) was lost owing to stopping the intervention. Usually, the effect of an intervention to reduce cardiovascular risk factors is lost soon after the cessation of the intervention. In some cases, however, the effect of a transient intervention on the control of risk factors significantly persisted, even during the post-trial observational period, with the significant suppression of the outcome. We defined the prolonged effect of the intervention as “carry-over effect” and distinguished it from “memory phenomenon”.
Twenty-one articles were found to describe the “memory phenomenon” as positive, after careful examination of the full-text of the 907 articles retrieved from the initial screening. Only three articles were judged as describing the “memory phenomenon” as negative. Five articles showed “carry-over effects” of the intervention (Table 1). The systematic review revealed that each intervention type, that is, glucose lowering, blood pressure lowering, or LDL-cholesterol lowering, possessed unique characteristics of the “memory phenomenon” (details are described in our report [20]). Most of the observational periods of these studies lasted for > 10 years. Therefore, “memory phenomenon” was suggested to last for a long time and is thought to have a considerable effect on the clinical course of NCDs [21,22,23].
The observation of “organ memory” in clinical settings and animal studies leads us to believe that “organ memory” participates in an incessant “updating” process provoked by external stimuli loaded on the organ. Memory is not merely an engraved mark that lacks elasticity and is unidirectional but originally contains multi-faceted phenomena, including “learning”, “imprinting”, “forgetting”, “recalling”, “erasing”, and “rewriting”. In the field of neuroscience, recent behavioral studies as well as molecular mechanistic studies have revealed that a series of “updating” processes is indispensable in maintaining neuronal memory [32, 33]. In other words, biological phenomena that exhibit the updating process can be referred to as “memory”. The “updating” of neuronal memory is completed through three processes, namely, 1. “recalling” of the memory, 2. “erasing” of the original memory, and 3. “rewriting” it with a new version of the memory.
Organs cannot possess “consciousness”, so it might not be appropriate to consider whether a “recalling” process exists in organs. However, the properties of organs are incessantly altered by external stimuli loaded on organs as if it is “updating”. Thus, “organ memory” should be distinctive from an existing concept of “organ remodeling” or “irreversible deviation”, because it is continuously “updating” through dynamic adaptive process owing to the repetitive external stimuli loaded on organs. For example, when repetitive exogenous noxious stimuli cause DNA damage in the cells of organs, they not only induce the DNA repair process each time but also influence the epigenetic status of DNA, which contributes to the long-term alteration of gene activation in organs (described in detail later). Importantly, epigenetic status is variously modulated, depending upon the kinds, severity, duration, and intervals of the stimuli. The precise relation of DNA repair and epigenetic modulation is explained in a recent review, and this process is analogous to neuronal memory “updating” [34].
It is clinically important to investigate whether “organ memory” can be “updated” by our behaviors. Possible manipulation of “organ memory” has been demonstrated in recent reports on cardiovascular diseases. Salt memory for hypertension has been erased by combining treatment with a calcium channel blocker and an angiotensin receptor blocker [19]. Established hypertensive nephrosclerosis and arteriosclerosis were also reversed by transient intensive RAS inhibition [35, 36]. The transient blockade of RAS reversed established glomerulosclerosis caused by Adriamycin administration [37]. In recent parabiosis (the joining of two animals) experiments using an older mouse and a younger mouse, rejuvenation occurred in the older mouse [38, 39]. That is, aging phenotypes (cardiomegaly, muscle fibrosis, and cognitive decline) observed in older mice were reported to recover as if reverting to a young state. Therefore, the modulation of humoral factors has the potential to update “organ memory”. In the clinical field, it has been reported that glomerular expansion was reduced and advanced stage diabetic nephropathy regressed owing to the transplantation of a healthy pancreas into a patient with type 1 diabetes [40]. We have also demonstrated that the alteration of the clinical course of hypertension was partly achievable by inhibiting RAS transiently (STAR CAST study) [41]. These results indicate that “organ memory” can be “updated”. The manipulation of “organ memory” is a worthwhile challenge that could be a new medical approach for NCDs.
As described above, even if the same stress is loaded on an organ, the organ response to the stress would differ in extent, speed, or direction from the previous response. Depending on the stress burden, the organ undergoes continuous remodeling of its structure and function to adapt to the stimulation. Therefore, after being subjected to a transient stress, the components of the organ shift to a different state/dimension, which arouses a different stress response. This adaptive time-dependent change in the organ stress responsiveness is “organ memory”. Then, the next question is “What is the mechanistic basis for “organ memory”?
It is becoming clear that many biological systems are governed by non-linear processes that are regulated by positive feedback mechanisms. These processes may switch between discrete states based on a functional relationship with the memory of input stimuli. The concept of “hysteresis” has been used to describe such memory-dependent non-linear processes in material sciences and has recently been applied to biological processes [42]. Cell fate commitment during oocyte maturation is a known example of such process that is governed by “hysteresis”. In oocytes, mitogen-activated protein kinase and cell-division cycle protein kinase Cdc2 are organized into positive feedback loops. Such time-dependent positive feedback mechanisms could produce an actively maintained “memory” and could explain the irreversibility of maturation [43]. In these contexts, we propose that “organ memory” is incessantly updated by transient stress loaded on organs, which results in the production of different organ responses in a spiral fashion (“the spiral model”, Fig. 2), driven by positive feedback mechanisms. We hypothesize that “organ memory” has a nature that is governed by “hysteresis” and repeated stimuli would designate the organ to a certain different state, which may lead to the occurrence and progression of diseases.
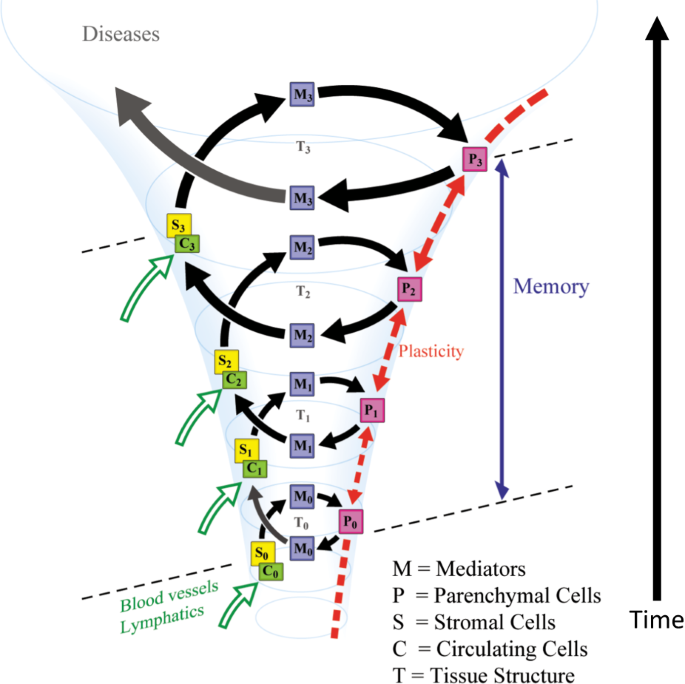
Repeated stimuli can continuously change the status of an organ through the signal networks governed by positive feedback loops. The multiple factors in the organ would consist of such networks that generate “organ memory” in a spiral fashion. Within the organ, the intricate cellular interactions of organ parenchymal cells, interstitial cells, and circulating cells that are delivered through blood and lymph vessels are postulated to participate in the generation of “organ memory.” Intercellular mediators, including hormones, cytokines, micro RNA, exosomes, and so on, also play significant roles in cellular communications that are crucial for the generation of “organ memory”. Alteration in tissue structure, such as the modification of the extracellular matrix through the generation of Amadori products due to hyperglycemia in diabetes mellitus, should affect “organ memory” [44]. Intracellular events, including the remodeling of organelle dynamics and metabolic processes and epigenetic modifications of gene expression, should play essential roles in the generation of “organ memory”.
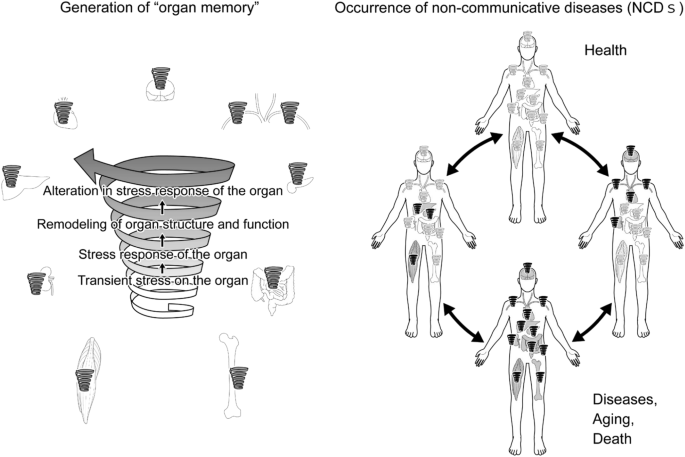
Once “organ memory” is established in an organ, “organ memory” in each organ can influence one another and affect “organ memory” in a different organ. We assume that organ failure that occurs in NCDs may develop in such inter-organ communication of “organ memory”, as demonstrated in Fig. 3.
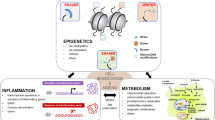
One of the significant elements responsible for “organ memory” is the epigenetic regulation of gene expression (Fig. 4). In the process of neuronal memory formation, epigenetic mechanisms have been proven to play an important role [45]. In the case of pro-inflammatory activation of cardiac fibroblasts, epigenetic changes in histone acetylation have been suggested to memorize a stimulation by angiotensin II [46]. For “angiotensin block memory”, which we originally reported as an organ memory, actual epigenetic changes occurred as a key step in the formation of memory in cases of human glomerulopathy, including diabetic nephropathy [47]. We demonstrated that the expression of the transcription factor, Kruppel-like Factor 4 (KLF4) was reduced in podocytes in patients with glomerular diseases. The reduced expression of KLF4 in podocytes leads to an increased methylation level of the nephrin promoter and decreased nephrin expression, resulting in proteinuria. Concerning the mechanism of the epigenetic regulation of the nephrin promoter, overexpression of KLF4 reduced the binding of DNA methyltransferase 1 to the nephrin promoter [48]. We further reported that angiotensin II, a crucial mediator of kidney diseases, suppressed KLF4 expression to modulate the long-term epigenetic status of the nephrin gene [37].
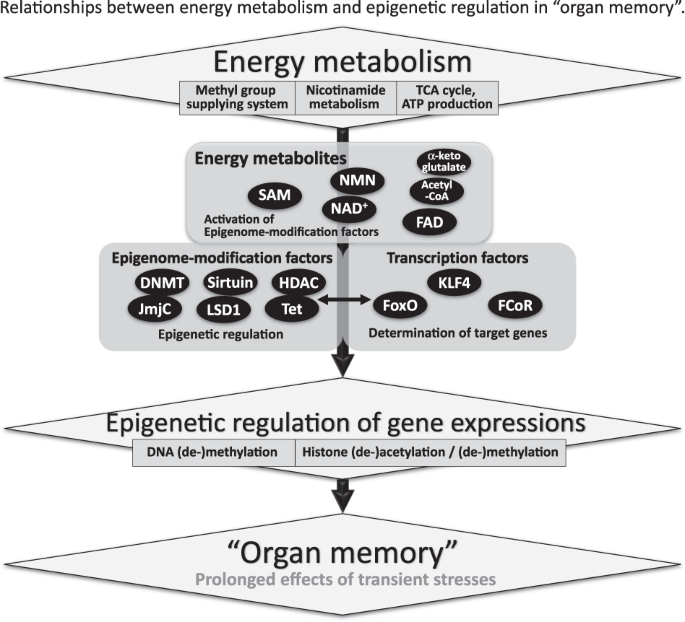
In these ways, both epigenome-modification enzymes, such as histone deacetylases and DNA methyltransferases, and transcription factors seem to be essential for the epigenetic regulation of gene expression, which is involved in the generation of “organ memory” (Fig. 4) [49]. Forkhead box O1 (FoxO1) is a transcription factor that can directly bind to the nicotinamide adenine dinucleotide (NAD)-dependent histone-deacetylase, Sirt1 [50]. Interactions between Sirt1, FoxO1, and the FoxO1-corepressor, which we identified and that represses activity of FoxO1 [51], are another example of the complex of transcription factors and epigenome-modification factors that would be involved in the generation of “organ memory”.
We also focused on the fact that “energy metabolites” with low molecular weights, such as NAD, flavin adenine dinucleotide, acetyl-CoA, and S-adenosylmethionine (SAM), are essential for epigenetic regulation (Fig. 4) [52]. These energy metabolites have been shown to regulate the activity of epigenome-modification factors, including sirtuins, histone deacetylases, DNMTs, and jumonji C. Recent reports have demonstrated that epigenetic modulation through these small molecule metabolites is involved in the progression of various diseases [53].
Several reports have demonstrated that dysregulation of nicotinamide metabolism causes widespread changes in the epigenetic regulation of gene expression. We reported that the activity of the histone-deacetylase Sirt1 in podocytes in patients with diabetes was reduced owing to an impairment in the transportation of the NAD precursor nicotinamide mononucleotide from the renal proximal tubules to the podocytes. Consequently, the expression of claudin-1 in podocytes was epigenetically upregulated, which led to impaired glomerular barrier function and albuminuria [54]. Nicotinamide N-methyltransferase (NNMT) is an enzyme that methylates nicotinamide using SAM as a methyl donor. Overexpression of NNMT has been shown to consume a large amount of SAM and impair cellular methylation potential. Conversely, NNMT inhibition increases cellular SAM levels, promotes methylation of histone H3K4 and, thereby, augments gene expression. The genetic depletion of NNMT in white adipose tissue and the liver has been reported to protect against diet-induced obesity in mice by upregulating the expression of genes related to energy expenditure through histone H3K4 methylation [55]. In cancer cells, overexpression of NNMT has been shown to contribute to tumorigenesis by decreasing histone methylation, which alters the expression of protumorigenic genes [56]. In these ways, cellular metabolism can epigenetically modulate the expression of genes that are related to the progression of diseases.
Transient changes in organ cellular metabolism can produce prolonged effects on the organ through the epigenetic regulation of gene expression. Transient hyperglycemia has been shown to induce long-lasting epigenetic changes in the promoter of the nuclear factor κB p65 subunit gene in cultured aortic endothelial cells, which resulted in the promotion of monocyte recruitment [57]. After completion of the DCCT/EDIC study [5, 6], the extent of H3K9 acetylation/methylation in monocytes of the participants was revealed to relate to diabetic vascular complications (“glucose memory”) [58]. The elucidation of these interactions between cellular metabolism and epigenetics would contribute to the further understanding of “organ memory”.
In the present review, we proposed that “organ memory” is another concept of memory, in addition to two types of “memory” that are recognized in brain and immune sciences. Future research on “organ memory” could be expanded in many directions, including the following approaches that would be effective in suppressing or regressing the chronic progression of NCDs, including hypertension and its organ complications.
These kinds of research approaches regarding “organ memory” could result in new treatments for NCDs and generate a new dimension of medical science.
We express sincere gratitude to Dr. Masami Tanaka (Keio University) for helpful contributions to this article.